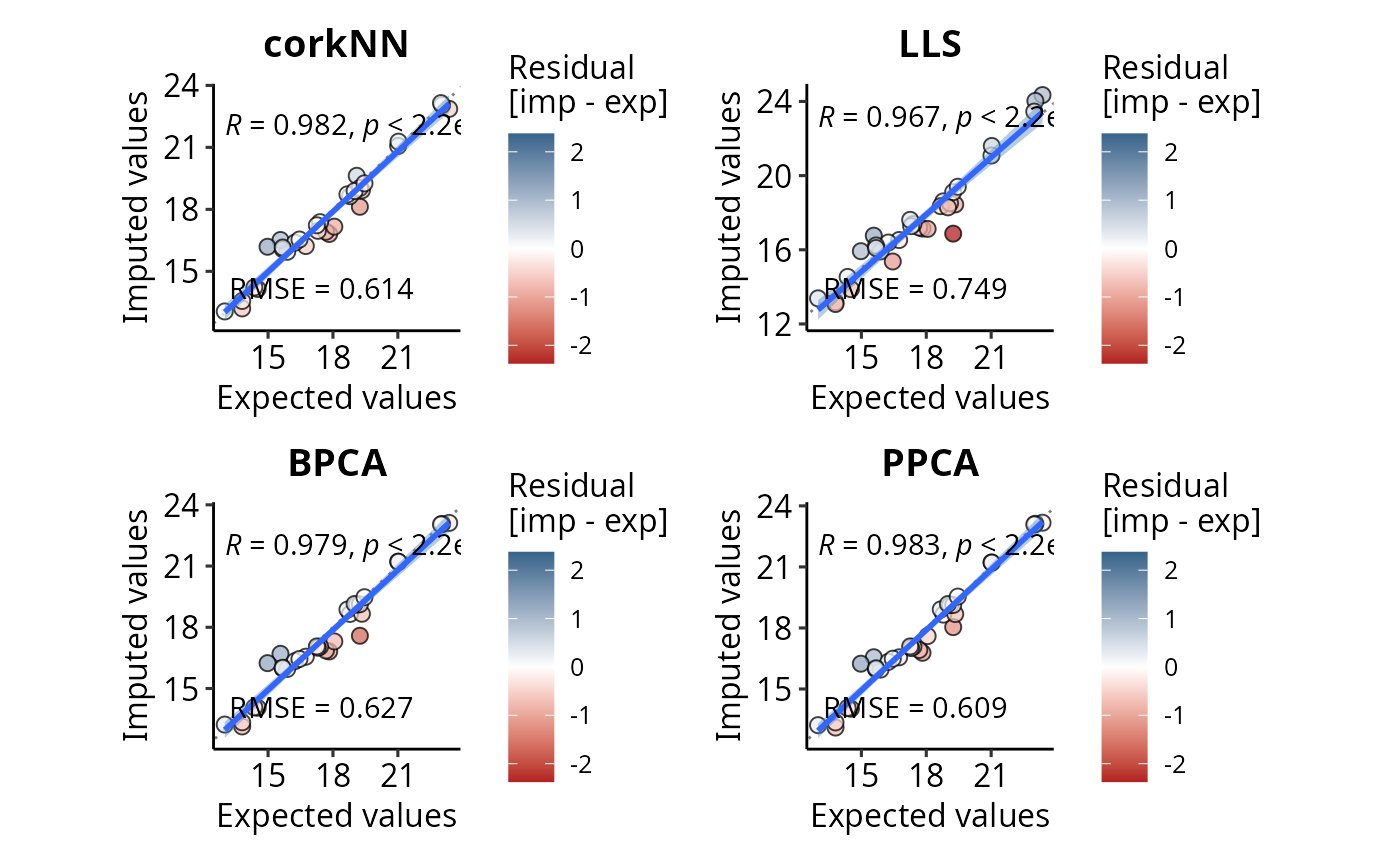
Function that computes the Root Mean Squared Error (RMSE) for all the 4 possibile imputation algorithms: missForest, kNN, LLS, SVD. A new dataset containing only proteins with known values is created, a certain percentage of NAs is then manually introduced (with the possibility to respect the "pattern" of the missing values). This percentage is equivalent to the percentage of missing values in the original data set. At the end the imputed values in the new data set are compared with the measured (expected) ones.
# S3 method for class 'imp.methods'
compare(
DEprot.object,
percentage.test = 30,
correlation.method = "pearson",
sample.group.column = NULL,
use.normalized.data = TRUE,
run.missForest = TRUE,
run.kNN = TRUE,
run.tkNN = TRUE,
run.corkNN = TRUE,
run.LLS = TRUE,
run.SVD = TRUE,
run.BPCA = TRUE,
run.PPCA = TRUE,
run.RegImpute = TRUE,
missForest.max.iterations = 100,
missForest.variable.wise.OOBerror = TRUE,
missForest.cores = 1,
missForest.parallel.mode = "variables",
kNN.n.nearest.neighbours = 10,
LLS.k = 2,
pcaMethods.nPCs.to.test = 5,
RegImpute.max.iterations = 10,
RegImpute.fillmethod = "row_mean",
normalize.color.bar = TRUE,
low.residual.color = "firebrick",
zero.residual.color = "white",
high.residual.color = "steelblue4",
seed = NULL,
verbose = FALSE
)Arguments
- DEprot.object
A
DEprot object, as generated by load.counts or load.counts2.- percentage.test
Numeric value between 0 (excluded) and 100 indicating the percentage of proteins to use for the test dataset. Default:
30.- correlation.method
String indicating the method to use for the correlations. One among: 'pearson', 'spearman'. Default:
"pearson".- sample.group.column
String indicating the ID of any column of the metadata table. This will be used to introduce the same frequencies of n-missing values for a protein and therefore not introducing the NAs completely at random in the dataset. Default:
NULL, NAs are assigned randomly (same percentage of NAs present in the original table).- use.normalized.data
Logical value indicating whether the imputation should be performed based on the rationalized data. Default:
TRUE.- run.missForest
Logical values indicating whether the test for the
missForestimputation should be performed. Default:TRUE.- run.kNN
Logical values indicating whether the test for the
kNNimputation method should be performed. Default:TRUE.- run.tkNN
Logical values indicating whether the test for the
tkNNimputation method should be performed. Default:TRUE.- run.corkNN
Logical values indicating whether the test for the
corkNNimputation method should be performed. Default:TRUE.- run.LLS
Logical values indicating whether the test for the
LLSimputation method should be performed. Default:TRUE.- run.SVD
Logical values indicating whether the test for the
SVDimputation method should be performed. Default:TRUE.- run.BPCA
Logical values indicating whether the test for the
BPCAimputation method should be performed. Default:TRUE.- run.PPCA
Logical values indicating whether the test for the
PPCAimputation method should be performed. Default:TRUE.- run.RegImpute
Logical values indicating whether the test for the
RegImputeimputation method should be performed. Default:TRUE.- missForest.max.iterations
Max number of iterations for the
missForestalgorithm. Default:100.- missForest.variable.wise.OOBerror
Logical value to define whether the OOB error is returned for each variable separately. Default:
TRUE.- missForest.cores
Number of cores used to run the
missForestalgorithm. IfmissForest.coresis 1 (or lower), the imputation will be run in parallel. Two modes are possible and can be defined by the parametermissForest.parallel.mode. Default:1.- missForest.parallel.mode
Define the mode to use for the parallelization, ignored when
coresis more than 1. One among: 'variables', 'forests'. Default:"variables". See also the documentation of the missForest function.- kNN.n.nearest.neighbours
Numeric value indicating the number of nearest neighbors to use to perform the
kNNimputation. Default:10.- LLS.k
Cluster size, this is the number of similar genes used for regression. Default:
2.- pcaMethods.nPCs.to.test
Numeric value indicating the number of Principal Components to test in order to find the optimal number of PCs to used in the imputation methods from the
pcaMethodspackage. This includes: 'LLS', 'SVD' (a.k.a 'svdImpute'), 'BPCA-pcaMethods', and 'PPCA'. Default:5.- RegImpute.max.iterations
Numeric value indicating the number of maximum iteration for the imputation with
RegImpute(fromDreamAI). Default:10.- RegImpute.fillmethod
String identifying the fill method to be used in the
RegImputemethod (fromDreamAI). One among"row_mean"and"zeros". Default:"row_mean". It throws an warning if"row_median"is used.- normalize.color.bar
Logical value indicating whether the color bar limits for the residuals in the correlation plots should be normalized among the methods. Default:
TRUE, the residual color bar absolute maximum is set to the max of all the residuals identified in all the methods,- low.residual.color
String indicating any R-supported color that must be used for the negative values of the residuals color bar in the correlation plots. Default:
"firebrick".- zero.residual.color
String indicating any R-supported color that must be used for the null residuals (zero, mid gradient color) color bar in the correlation plots. Default:
"white".- high.residual.color
String indicating any R-supported color that must be used for the positive values of the residuals color bar in the correlation plots. Default:
"steelblue4".- seed
Numeric value indicating the seed to use for the randomization. Default:
NULL, automatically generated (saved in theseedslot in the final object).- verbose
Logical valued indicating whether processing messages should be printed. Default:
FALSE.
Value
A DEprot.RMSE object.
See also
missForest, VIM, pcaMethods R-packages.
Examples
comparison <- compare.imp.methods(DEprot.object = DEprot::test.toolbox$dpo.norm,
percentage.test = 100,
sample.group.column = "combined.id",
run.missForest = FALSE,
run.BPCA = TRUE,
run.PPCA = TRUE,
run.kNN = FALSE,
run.tkNN = FALSE,
run.corkNN = TRUE,
run.LLS = TRUE,
run.SVD = FALSE,
run.RegImpute = FALSE)
#> Warning: The number of usable proteins is lower than the number of proteins requested.
summary(comparison)
#> imputation.method RMSE correlation.coeff processing.time rank
#> PPCA PPCA 0.6086783 0.9830473 0.39 1
#> corkNN corkNN 0.6144851 0.9824167 0.00 2
#> BPCA BPCA 0.6272062 0.9791381 0.62 3
#> LLS LLS 0.7489155 0.9674068 0.01 4
comparison