![]()

![]()

snakeATAC
- Introduction
- Installation an dependencies
- How to run the pipeline
- Results
- Package history and releases
- Contact
- License
Introduction
SnakeATAC is a snakemake based end-to-end pipeline to analyze ATAC-seq data. The input files required to run the pipeline are Paired-End fastq files. The pipeline include data quality check and normalization. It is included also a step of data reads shifting in order to take into account the Tn5 transposome insertion bias. Indeed, reads should be shifted +4bp and −5bp for positive and negative strand respectively, to account for the 9-bp duplication created by DNA repair of the nick by Tn5 transposase and achieve base-pair resolution of TF footprint and motif-related analyses (Yan F., et al., Genome Biol. 2020).
Citation
If you use this package, please cite:
Installation an dependencies
To install the pipeline it is required to download this repository and the installation of a conda environment is strongly suggested. Follow the following steps for the installation:
- Place yourself in the directory where the repository should be downloaded with
cd </target/folder> - download the GitHub repository with
git clone https://github.com/sebastian-gregoricchio/snakeATAC, or click on Code > Download ZIP on the GitHub page - install the conda environment from the yaml environment file contained in the repository:
conda env create -f </target/folder>/snakeATAC/workflow/envs/snakeATAC_conda_env_stable.yaml - activate the environment:
conda activate snakeATAC(if the env is not activated the pipeline won’t work)
How to run the pipeline
The snakemake pipeline requires only two files: a) the .snakefile, containing all the rules that will be run; b) the configuration.yaml file, in which the user can define and customize all the parameters for the different pipeline steps.
To partially avoid unexpected errors during the execution of the pipeline, a so called ‘dry-run’ is strongly recommended. Indeed, adding a -n at the end of the snakemake running command will allow snakemake to check that all links and file/parameters dependencies are satisfied before to run the “real” processes. This command will therefore help the debugging process.
snakemake -s </target/folder>/snakeATAC/workflow/snakeATAC.snakefile --configfile </target/folder>/snakeATAC/config/snakeATAC_config.yaml --cores 12 -n
If no errors occur, the pipeline can be run with the same command line without the terminal -n:
snakemake -s </target/folder>/snakeATAC/workflow/snakeATAC.snakefile --configfile </target/folder>/snakeATAC/config/snakeATAC_config.yaml --cores 12
Notice that the absence of errors does not mean that the pipeline will run without any issues; the “dry-run” is only checking whether all the resources are available.
Furthermore, there is the possibility to run the pipeline only partailly. An example of usage could be if someone wants to have a look to the fast quality controls (fastQC and multiQC reports) before to perform the alignment. To do that, it is sufficient to run a dry-run (-n mode); pick the name of the rule at which you want the pipeline to stop; type the following command:
snakemake -s </target/folder>/snakeATAC/workflow/snakeATAC.snakefile --configfile </target/folder>/snakeATAC/config/snakeATAC_config.yaml --cores 12 --until stop_rule_name
Snakefile
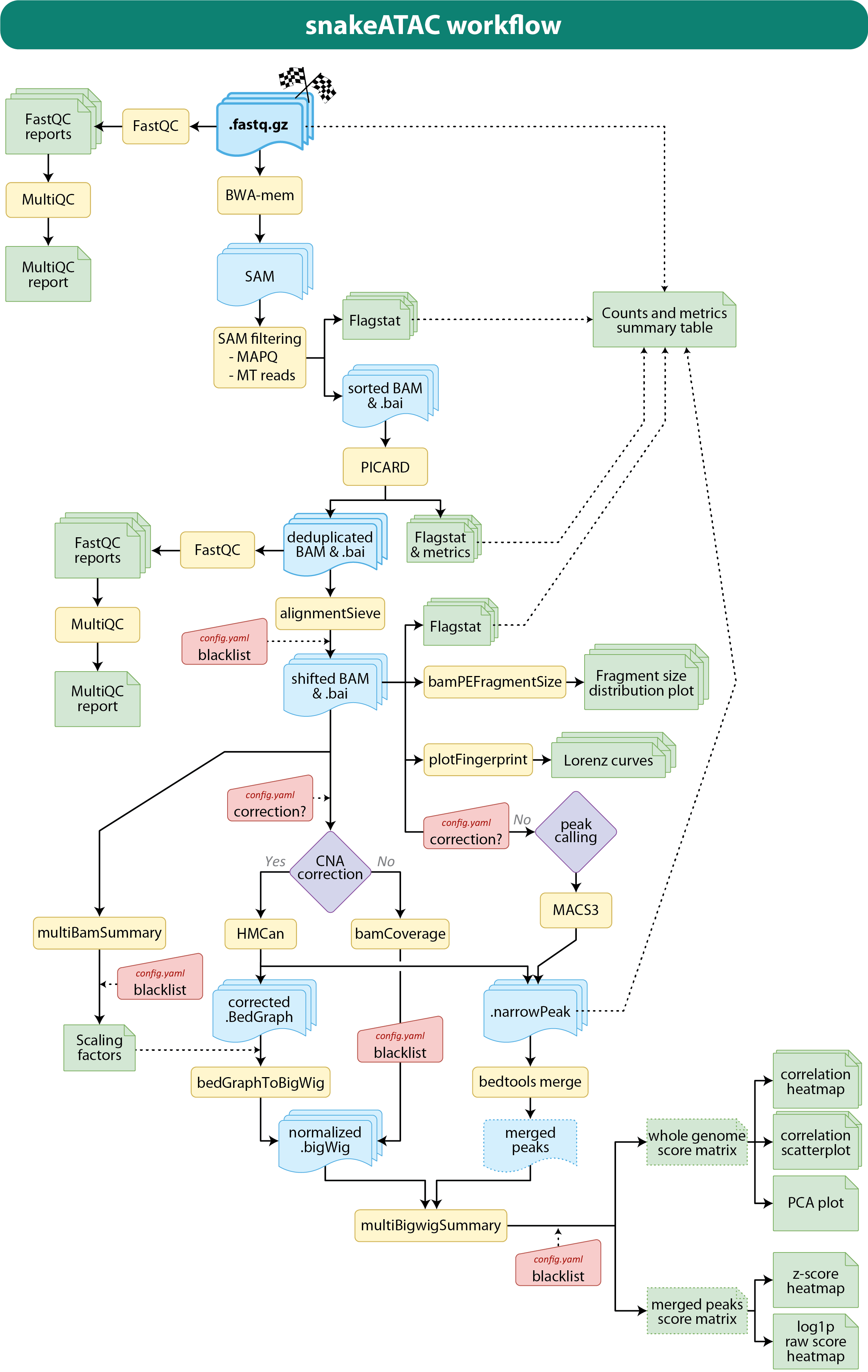
The snakefile are contained all the condition checks and rules (processing steps) that will be performed by the pipeline. In the following schematic mapping the main steps performed by the pipeline are depicted.
Briefly, first of a quality control (fastQC) of the raw fastq data is performed. Then, bwa-mem is used to align the paired-end sequences onto the genome of reference generating temporary SAM files that are further filter for mapping quality score and deprived of mitochondrial reads before to be converted to BAM. From this alignment files duplicated reads are removed (or marked) by PICARD and then shifted to correct the Tn5 nick repair bias (Yan F., et al., Genome Biol. 2020) by deeptools alignmentSieve tool. A quality control for each alignment and a multiQC report are generated, as well samtools flagstat metrics and deeptools fragment size distribution plots (see 03_BAM_dedup / 03_BAM_mdup paragraph for more details).
Then two possible path can be taken in order to normalize the data:
- HMcan corection (still in beta-test phase)
The sequencing signal can be corrected for Copy Number Variation (CNV); particularly useful for heterogeneous tumor samples. Then the corrected signal in bedGraph format is normalized on sequencing depth by multiplying the scores by the scaling factors computed by deeptools multiBamSummary. Finally, the normalized bedGraph files are converted to bigWig by the UCSC bedGraphToBigWig tool. HMcan will perfomr the peak calling and generate the narrowPeak files. - Classic method without CNV correction
Peaks are called on shifted bam files by MACS3 which will generate narrowPeak files. In parallel bam files are normalized by sequencing depth by deeptools bamCoverage. Independently of which method has been chosen, deeptools multiBigwigSummary will be used to generate a whole genome score matrix useful to infer the overall correlation/variability of the samples. This matrix is he input of deeptools plotCorrealtion and poltPCA (see 06_Overall_quality_and_info paragraph for more details).
Another analyses performed to determine sample variability is the quantification of the signal at any peak called in any sample. For the all the narrowPeak files are concatenated and merged together by bedtools merge and the score at each peak for each sample computed by multiBigwigSummary. The resulting matrix is used to generate two clustered heatmaps: one on the log1p of the raw scores, and one on the z-score (on rows).
More details on parameters and structure/meaning of the results can be found in the next paragraphs.
Configuration file
The configuration file is a yaml-formatted file containing all the parameters that are passed to different steps of the pipelines such as the directory with the input files, reference genome, threads of the tools, etc.
The snakeATAC configuration file is divided in two sections: a) ‘experiment-specific’, with al the parameters that most likely are changing from experiment to experiment; b) ‘common’, with parameters that are quite stable independently of the experiments design. The latter should be changed only for very specific needs and is in turn compose by two sections depending on whether the copy number variation is performed or not.
Hereafter, the meaning of the different parameters is described.
Experiment-specific section
| Parameter | Description |
|---|---|
| runs_directory | The full path to the directory were the input fastq files are contained, e.g. /home/user/ATAC/00_runs/. Importantly, the name of the files, deprived of the read suffix (e.g., _R1/_R2) and file extension (e.g., .fastq.gz) will be used as sample name. |
| output_directory | The full path to the folder in which the results should be stored, e.g. "/home/user/ATAC/". |
| fastq_extension | The extension of the input fastq files. This string will be removed from the input file names to obtain the sample names. Examples: ".fastq.gz", ".fq.gz", ".fasta", ".fq". |
| runs_suffix | A list (python format) with the two reads suffixes corresponding to the read1 and read2 for the paired-end sequencing. Example: ["_R1", "_R2"]. |
| blacklist_file | The full path to a BED-formatted file containing the regions corresponding to blacklisted regions (regions masked for normalization and peak calling). Blacklisted regions for the most common genome assemblies can be downloaded from the Boyle lab’s git-page. |
| genome_fasta | The full path to a fasta-formatted file containing the sequence of there reference genome into which the reads will be aligned. If the index (.fai) file is not present in the same folder, it will be build by the pipeline during its execution. The reference genomes can be downloaded, for instance, from the UCSC golden path. |
| perform_HMCan_correction | Default "False". If set to "True" the signal will be corrected for the presence of CNVs (Copy Number Variations) and the peak calling will be performed by HMCan. This correction could be useful in the case of heterogeneous cancer samples. Instead, if set to "False", the CNVs correction is skipped and the peak calling performed by MACS3. The CNV correction implementation is still in beta-testing phase. |
| effective_genomeSize | Effective genome size used to normalize the ATAC-seq signal; e.g. for Hg38: 2900338458. A table for the most common genome assemblies is available in this repository at snakeATAC/resources/chromosome_sizes_for_normalization.yaml. |
| ignore_for_normalization | A string of space separated chromosome names that should be excluded for ATAC-seq signal normalization, e.g. "X Y MT M KI270728.1". A table for the most common genome assemblies is available in this repository at snakeATAC/resources/chromosome_sizes_for_normalization.yaml. |
| call_variants | If true, variant calling by GATK4 will be performed. |
| call_SNPs | If true, Single Nucleotide Variation (SNP) calling by GATK4 will be performed. |
| call_indels | If true, Insertion/Deletion (indel) calling by GATK4 will be performed. |
| dbsnp_file | SNP database file (.dbsnp) for base recalibration required by GATK4. It could happen that your .bam files contain the ‘chr’ prefix in the chromosome names while your dbSNP file does not (or vice versa). This can be fixed in the .dbsnp file with the bcftools annotate --rename-chrs command. For Hg38, for instance, the dbSNP file can be downloaded from the broad institute cloud storage. Do not forget to download the INDEX as well! |
Common section
| Parameter | Description |
|---|---|
| fastQC_threads | Default: 2. Number of CPUs to use for fastQC (fastq quality control) |
| bwa_threads | Default: 8. Number of CPUs to use for the mapping performed by bwa-mem. |
| mapQ_cutoff | Default: 20. All reads with a mapping quality (MAPQ) score lower than this value will be filtered out from the bam files. |
| SAMtools_threads | Default: 8. Number of CPUs used by samtools for bam indexing and filtering. |
| remove_duplicates | Default: "true". Logical value to indicate whether the optical duplicates should be removed from the bams by PICARD. If set as "true" the suffix of output bam files will be ‘_dedup’ and the duplicates will be removed, otherwise the suffix will be ‘_mdup’ and the duplicates only marked when set to "false". |
| PICARD_max_records_in_ram | Default: 250000 This will specify the number of records stored in RAM before spilling to disk. The higher the number the higher the amount of RAM needed. |
| PICARD_max_file_handles_for_read_ends_map | Default: 4000 Maximum number of file handles to keep open when spilling read ends to disk. This number should be slightly lower than the per-process maximum number of file that may be open in your system (ulimit -n). |
| minFragmentLength | Default: 0. Minimum fragment length needed for pair inclusion during Tn5 tagmentation bias correction by deeptools alignmentSieve tool. |
| maxFragmentLength | Default: 0. Maximum fragment length needed for pair inclusion during Tn5 tagmentation bias correction by deeptools alignmentSieve tool. A value of 0 indicates no limit. |
| plot_format | Default: "pdf". File format of the images of the fragment size distribution generated by deeptools bamPEFragmentSize. |
| bamPEFragmentSize_threads | Default: 0. Number of CPUs deeptools bamPEFragmentSize should use to compute the fragment size distribution. |
| max_fragment_length | Default: 2000. Maximum fragment size length to be plotted by deeptools bamPEFragmentSize. A value of 0 indicates to use twice the mean fragment length. |
| window_length | Default: 1000. Size, in bp, of the sliding window used to compute the fragment size distribution by deeptools bamPEFragmentSize. |
| plotFingerprint_threads | Default: 8. Number of CPUs to be used by deeptools plotFingerprint to compute the Lorenz curves. |
| plotFingerprint_binSize | Default: 500. Size of the bins, in bp, by which the genome should be subdivided in order to compute the Lorenz curves by deeptools plotFingerprint. |
| plotFingerprint_sampledRegions | Default: 500000. Number of regions to be samples in order to compute the Lorenz curves by deeptools plotFingerprint. |
| plotFingerprint_extra_parameters | Default: "" (empty). A string with any additional parameter to add for the Lorenz curve computation performed by deeptools plotFingerprint. |
| binning_window_size | Default: 10000. Size of the bins, in bp, by which the genome should be subdivided in order to compute the average score matrix by deeptools multiBigwigSummary. This matrix is used to compute sample correlation and Principal Component Analyses (PCA). |
| multiBigwigSummary_threads | Default: 4. Number of CPUs to be used by deeptools multiBigwigSummary in order to compute an average signal over all the genome for each sample. The resulting matrix is used to compute sample correlation and Principal Component Analyses (PCA). |
| heatmap_color | Default: "Blues". A string indicating the color gradient pattern to use for the correlation heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: ‘Accent’, ‘Blues’, ‘BrBG’, ‘BuGn’, ‘BuPu’, ‘CMRmap’, ‘Dark2’, ‘GnBu’, ‘Greens’, ‘Greys’, ‘OrRd’, ‘Oranges’, ‘PRGn’, ‘Paired’, ‘Pastel1’, ‘Pastel2’, ‘PiYG’, ‘PuBu’, ‘PuBuGn’, ‘PuOr’, ‘PuRd’, ‘Purples’, ‘RdBu’, ‘RdGy’, ‘RdPu’, ‘RdYlBu’, ‘RdYlGn’, ‘Reds’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Spectral’, ‘Wistia’, ‘YlGn’, ‘YlGnBu’, ‘YlOrBr’, ‘YlOrRd’, ‘afmhot’, ‘autumn’, ‘binary’, ‘bone’, ‘brg’, ‘bwr’, ‘cividis’, ‘cool’, ‘coolwarm’, ‘copper’, ‘cubehelix’, ‘flag’, ‘gist_earth’, ‘gist_gray’, ‘gist_heat’, ‘gist_ncar’, ‘gist_rainbow’, ‘gist_stern’, ‘gist_yarg’, ‘gnuplot’, ‘gnuplot2’, ‘gray’, ‘hot’, ‘hsv’, ‘icefire’, ‘inferno’, ‘jet’, ‘magma’, ‘mako’, ‘nipy_spectral’, ‘ocean’, ‘pink’, ‘plasma’, ‘prism’, ‘rainbow’, ‘rocket’, ‘seismic’, ‘spring’, ‘summer’, ‘tab10’, ‘tab20’, ‘tab20b’, ‘tab20c’, ‘terrain’, ‘twilight’, ‘twilight_shifted’, ‘viridis’, ‘vlag’, ‘winter’. |
| zScore_heatmap_color | Default: "seismic". A string indicating the color gradient pattern to use for the peak score heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: ‘Accent’, ‘Blues’, ‘BrBG’, ‘BuGn’, ‘BuPu’, ‘CMRmap’, ‘Dark2’, ‘GnBu’, ‘Greens’, ‘Greys’, ‘OrRd’, ‘Oranges’, ‘PRGn’, ‘Paired’, ‘Pastel1’, ‘Pastel2’, ‘PiYG’, ‘PuBu’, ‘PuBuGn’, ‘PuOr’, ‘PuRd’, ‘Purples’, ‘RdBu’, ‘RdGy’, ‘RdPu’, ‘RdYlBu’, ‘RdYlGn’, ‘Reds’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Spectral’, ‘Wistia’, ‘YlGn’, ‘YlGnBu’, ‘YlOrBr’, ‘YlOrRd’, ‘afmhot’, ‘autumn’, ‘binary’, ‘bone’, ‘brg’, ‘bwr’, ‘cividis’, ‘cool’, ‘coolwarm’, ‘copper’, ‘cubehelix’, ‘flag’, ‘gist_earth’, ‘gist_gray’, ‘gist_heat’, ‘gist_ncar’, ‘gist_rainbow’, ‘gist_stern’, ‘gist_yarg’, ‘gnuplot’, ‘gnuplot2’, ‘gray’, ‘hot’, ‘hsv’, ‘icefire’, ‘inferno’, ‘jet’, ‘magma’, ‘mako’, ‘nipy_spectral’, ‘ocean’, ‘pink’, ‘plasma’, ‘prism’, ‘rainbow’, ‘rocket’, ‘seismic’, ‘spring’, ‘summer’, ‘tab10’, ‘tab20’, ‘tab20b’, ‘tab20c’, ‘terrain’, ‘twilight’, ‘twilight_shifted’, ‘viridis’, ‘vlag’, ‘winter’. |
Copy Number Variation signal correction (for details see the HMCan page)
| Parameter | Description |
|---|---|
| HMCan_path | Full path to the folder containing the HMCan scripts. Instruction for the download and build of HMCan can be found at the the HMCan page. |
| HMCan_threads | Default: 12. Number of CPUs to be used by HMCan. |
| chromosome_sizes_file | Full path to the chromosome sizes file used to convert the CNV corrected signal into a bigWig. These files can be downloaded from the UCSC golden path. |
| reference_sample | Default: "NA". A string to define the reference sample (sample ID) to which all the samples should be normalized. "NA" indicates that the first samples in alphabetic order will be used as reference. |
| format | Default: "BAM" (used to build an HMCan configFile). |
| GCIndex | (used to build an HMCan configFile) Full path to the GC index file provided at the HMCan page. Example: “/home/user/HMCan-master/data/GC_profile_25KbWindow_Mapp100bp_hg38.cnp”. |
| smallBinLength | Default: 5 (used to build an HMCan configFile). For details see the HMCan page. |
| largeBinLength | Default: 25000 (used to build an HMCan configFile). For details see the HMCan page. |
| *pvalueThreshold | Default: 0.001 (used to build an HMCan configFile). For details see the HMCan page. |
| mergeDistance | Default: 0 (used to build an HMCan configFile). For details see the HMCan page. |
| iterationThreshold | Default: 5 (used to build an HMCan configFile). For details see the HMCan page. |
| finalThreshold | Default: 0 (used to build an HMCan configFile). For details see the HMCan page. |
| maxIter | Default: 20 (used to build an HMCan configFile). For details see the HMCan page. |
| PrintWig | Default: "False" (used to build an HMCan configFile). For details see the HMCan page. |
| PrintPosterior | Default: "True" (used to build an HMCan configFile). For details see the HMCan page. |
| PrintBedGraph | Default: "True" (used to build an HMCan configFile). For details see the HMCan page. |
| CallPeaks | Default: "True" (used to build an HMCan configFile). For details see the HMCan page. |
| pairedEnds | Default: "True" (used to build an HMCan configFile). For details see the HMCan page. |
| Type | Default: "ATAC-seq" (used to build an HMCan configFile). For details see the HMCan page. |
| GCmergeDistance | Default: 1000 (used to build an HMCan configFile). For details see the HMCan page. |
| RemoveDuplicates | Default: "False" (used to build an HMCan configFile). For details see the HMCan page. |
| CNAnormalization | Default: "True" (used to build an HMCan configFile). For details see the HMCan page. |
| multiBamSummary_threads | Default: 6. Number of CPUs to be used by deeptools multiBamSummary in order to compute an average signal over all the genome for each sample for the calculation of the scaling factors. This factors will be used to normalize the CNV corrected signal by sequencing depth. |
Standard normalization and peak calling (without CNV correction)
| Parameter | Description |
|---|---|
| bigWig_binSize | Default: 5. Size, in bp, of the bins used to compute the normalized bigWig files. |
| normalization_method | Default: "RPGC", reads per genomic content (1x normalization). Type of normalization to be used to generated the normalized bigWig files by deeptools bamCoverage. |
| bamCoverage_threads | Default: 8. Number of CPUs to be used by deeptools bamCoverage in order to compute the signal normalization and the bigWig generation. |
| genome_size_MACS | A string or a number indicating the genome size to use by MACS3 for the peak calling. Example: "hs". Some values: hs = 2.7e9, mm = 1.87e9, ce = 9e7, dm = 1.2e8. |
| FDR_cutoff | Deafult: 0.01. False Discovery Ratio (FDR) cutoff used by MACS3 to filter the significant peaks. |
| call_summits | Default: "True". Logical value to define whether MACS3 should also call the peak summits (position with the highest value). |
| FRiP_threshold | Default 20. This value will be used to label the FRiP (Fraction of Reads in a Peak) score as “good” or “bad in the summary table for each single sample. A FRiP above the 20% (FRiP = 0.02) is considered a good score for ATAC-seq peaks by the ENCODE guidelines. |
Samples comparison for overall variance estimation
| Parameter | Description |
|---|---|
| binning_window_size | Default: 10000 (base-pairs). Size of the window used to scan the genome and calculate the bins average score for the sample correlation. |
| multiBigwigSummary_threads | Default: 4. Number of threads to be used by multiBigwigSummary (deeptools) to compute the matrix for the sample peak score correlation. |
| heatmap_color | Default: "Blues". A string indicating the color gradient pattern to use for the peaks score correlation heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: ‘Accent’, ‘Blues’, ‘BrBG’, ‘BuGn’, ‘BuPu’, ‘CMRmap’, ‘Dark2’, ‘GnBu’, ‘Greens’, ‘Greys’, ‘OrRd’, ‘Oranges’, ‘PRGn’, ‘Paired’, ‘Pastel1’, ‘Pastel2’, ‘PiYG’, ‘PuBu’, ‘PuBuGn’, ‘PuOr’, ‘PuRd’, ‘Purples’, ‘RdBu’, ‘RdGy’, ‘RdPu’, ‘RdYlBu’, ‘RdYlGn’, ‘Reds’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Spectral’, ‘Wistia’, ‘YlGn’, ‘YlGnBu’, ‘YlOrBr’, ‘YlOrRd’, ‘afmhot’, ‘autumn’, ‘binary’, ‘bone’, ‘brg’, ‘bwr’, ‘cividis’, ‘cool’, ‘coolwarm’, ‘copper’, ‘cubehelix’, ‘flag’, ‘gist_earth’, ‘gist_gray’, ‘gist_heat’, ‘gist_ncar’, ‘gist_rainbow’, ‘gist_stern’, ‘gist_yarg’, ‘gnuplot’, ‘gnuplot2’, ‘gray’, ‘hot’, ‘hsv’, ‘icefire’, ‘inferno’, ‘jet’, ‘magma’, ‘mako’, ‘nipy_spectral’, ‘ocean’, ‘pink’, ‘plasma’, ‘prism’, ‘rainbow’, ‘rocket’, ‘seismic’, ‘spring’, ‘summer’, ‘tab10’, ‘tab20’, ‘tab20b’, ‘tab20c’, ‘terrain’, ‘twilight’, ‘twilight_shifted’, ‘viridis’, ‘vlag’, ‘winter’. |
| zScore_heatmap_color | Default: "seismic". A string indicating the color gradient pattern to use for the peak score heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: ‘Accent’, ‘Blues’, ‘BrBG’, ‘BuGn’, ‘BuPu’, ‘CMRmap’, ‘Dark2’, ‘GnBu’, ‘Greens’, ‘Greys’, ‘OrRd’, ‘Oranges’, ‘PRGn’, ‘Paired’, ‘Pastel1’, ‘Pastel2’, ‘PiYG’, ‘PuBu’, ‘PuBuGn’, ‘PuOr’, ‘PuRd’, ‘Purples’, ‘RdBu’, ‘RdGy’, ‘RdPu’, ‘RdYlBu’, ‘RdYlGn’, ‘Reds’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Spectral’, ‘Wistia’, ‘YlGn’, ‘YlGnBu’, ‘YlOrBr’, ‘YlOrRd’, ‘afmhot’, ‘autumn’, ‘binary’, ‘bone’, ‘brg’, ‘bwr’, ‘cividis’, ‘cool’, ‘coolwarm’, ‘copper’, ‘cubehelix’, ‘flag’, ‘gist_earth’, ‘gist_gray’, ‘gist_heat’, ‘gist_ncar’, ‘gist_rainbow’, ‘gist_stern’, ‘gist_yarg’, ‘gnuplot’, ‘gnuplot2’, ‘gray’, ‘hot’, ‘hsv’, ‘icefire’, ‘inferno’, ‘jet’, ‘magma’, ‘mako’, ‘nipy_spectral’, ‘ocean’, ‘pink’, ‘plasma’, ‘prism’, ‘rainbow’, ‘rocket’, ‘seismic’, ‘spring’, ‘summer’, ‘tab10’, ‘tab20’, ‘tab20b’, ‘tab20c’, ‘terrain’, ‘twilight’, ‘twilight_shifted’, ‘viridis’, ‘vlag’, ‘winter’. |
| zScore_heatmap_color | Default: "seismic". A string indicating the color gradient pattern to use for the peak score heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: ‘Accent’, ‘Blues’, ‘BrBG’, ‘BuGn’, ‘BuPu’, ‘CMRmap’, ‘Dark2’, ‘GnBu’, ‘Greens’, ‘Greys’, ‘OrRd’, ‘Oranges’, ‘PRGn’, ‘Paired’, ‘Pastel1’, ‘Pastel2’, ‘PiYG’, ‘PuBu’, ‘PuBuGn’, ‘PuOr’, ‘PuRd’, ‘Purples’, ‘RdBu’, ‘RdGy’, ‘RdPu’, ‘RdYlBu’, ‘RdYlGn’, ‘Reds’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Spectral’, ‘Wistia’, ‘YlGn’, ‘YlGnBu’, ‘YlOrBr’, ‘YlOrRd’, ‘afmhot’, ‘autumn’, ‘binary’, ‘bone’, ‘brg’, ‘bwr’, ‘cividis’, ‘cool’, ‘coolwarm’, ‘copper’, ‘cubehelix’, ‘flag’, ‘gist_earth’, ‘gist_gray’, ‘gist_heat’, ‘gist_ncar’, ‘gist_rainbow’, ‘gist_stern’, ‘gist_yarg’, ‘gnuplot’, ‘gnuplot2’, ‘gray’, ‘hot’, ‘hsv’, ‘icefire’, ‘inferno’, ‘jet’, ‘magma’, ‘mako’, ‘nipy_spectral’, ‘ocean’, ‘pink’, ‘plasma’, ‘prism’, ‘rainbow’, ‘rocket’, ‘seismic’, ‘spring’, ‘summer’, ‘tab10’, ‘tab20’, ‘tab20b’, ‘tab20c’, ‘terrain’, ‘twilight’, ‘twilight_shifted’, ‘viridis’, ‘vlag’, ‘winter’. |
| zScore_heatmap_color | Default: "seismic". A string indicating the color gradient pattern to use for the peak score heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: ‘Accent’, ‘Blues’, ‘BrBG’, ‘BuGn’, ‘BuPu’, ‘CMRmap’, ‘Dark2’, ‘GnBu’, ‘Greens’, ‘Greys’, ‘OrRd’, ‘Oranges’, ‘PRGn’, ‘Paired’, ‘Pastel1’, ‘Pastel2’, ‘PiYG’, ‘PuBu’, ‘PuBuGn’, ‘PuOr’, ‘PuRd’, ‘Purples’, ‘RdBu’, ‘RdGy’, ‘RdPu’, ‘RdYlBu’, ‘RdYlGn’, ‘Reds’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Spectral’, ‘Wistia’, ‘YlGn’, ‘YlGnBu’, ‘YlOrBr’, ‘YlOrRd’, ‘afmhot’, ‘autumn’, ‘binary’, ‘bone’, ‘brg’, ‘bwr’, ‘cividis’, ‘cool’, ‘coolwarm’, ‘copper’, ‘cubehelix’, ‘flag’, ‘gist_earth’, ‘gist_gray’, ‘gist_heat’, ‘gist_ncar’, ‘gist_rainbow’, ‘gist_stern’, ‘gist_yarg’, ‘gnuplot’, ‘gnuplot2’, ‘gray’, ‘hot’, ‘hsv’, ‘icefire’, ‘inferno’, ‘jet’, ‘magma’, ‘mako’, ‘nipy_spectral’, ‘ocean’, ‘pink’, ‘plasma’, ‘prism’, ‘rainbow’, ‘rocket’, ‘seismic’, ‘spring’, ‘summer’, ‘tab10’, ‘tab20’, ‘tab20b’, ‘tab20c’, ‘terrain’, ‘twilight’, ‘twilight_shifted’, ‘viridis’, ‘vlag’, ‘winter’. |
GATK variant calling
| Parameter | Description |
|---|---|
| bgzip_threads | Default: 4. Number of threads to be used by samtools bgzip to compress VCFs (Variant Call Format). |
| DP_snp_threshold | Default: 20. Threshold for the hard-filtering of the SNP (single-nucleotide polymorphism) VCF table for sequencing depth (DP) by SnpSift Filter. |
| QUAL_snp_threshold | Default: 0. Threshold for the hard-filtering of the SNP (single-nucleotide polymorphism) VCF table for quality (QUAL) by SnpSift Filter. |
| DP_indel_threshold | Default: 20. Threshold for the hard-filtering of the InDel (insertion/delition) VCF table for sequencing depth (DP) by SnpSift Filter. |
| QUAL_indel_threshold | Default: 0. Threshold for the hard-filtering of the InDel (insertion/delition) VCF table for quality (QUAL) by SnpSift Filter. |
| SnpSift_vcf_fields_to_extract | Default: [ "CHROM", "POS", "ID", "REF", "ALT", "QUAL", "DP", "FILTER", "FORMAT", "GEN[*].GT", "GEN[*].AD" ]. A python list to indicate the fields to be extracted from the VCFs (both SNP and InDel) during the conversion to .txt file by SnpSift Extract Fields. |
Results
The structure of the output_folder is the following:
output_folder
├── 01_fastQC_raw
│ ├── sample_fastqc.html
│ ├── sample_fastqc.zip
│ └── multiQC_raw
│ ├── multiQC_report_fastqRaw_data
│ │ ├── multiqc_citations.txt
│ │ ├── multiqc_data.json
│ │ ├── multiqc_fastqc.txt
│ │ ├── multiqc_general_stats.txt
│ │ ├── multiqc.log
│ │ └── multiqc_sources.txt
│ └── multiQC_report_fastqRaw.html
|
├── 02_BAM
│ ├── sample_mapQ20_sorted_woMT.bam
│ ├── sample_mapQ20_sorted_woMT.bam.bai
│ └── flagstat
│ ├── sample_flagstat_filtered_bam_woMT.txt
│ └── sample_flagstat_UNfiltered_bam.txt
|
├── 03_BAM_dedup (or 03_BAM_mdup, if duplicates are not removed)
│ ├── sample_mapQ20_woMT_dedup_shifted_sorted.bam
│ ├── sample_mapQ20_woMT_dedup_shifted_sorted.bam.bai
│ ├── fastQC
│ │ ├── sample_sorted_woMT_dedup_fastqc.html
│ │ ├── sample_sorted_woMT_dedup_fastqc.zip
│ │ └── multiQC_dedup_bams
│ │ ├── multiQC_report_BAMs_dedup_data
│ │ │ ├── multiqc_citations.txt
│ │ │ ├── multiqc_data.json
│ │ │ ├── multiqc_fastqc.txt
│ │ │ ├── multiqc_general_stats.txt
│ │ │ ├── multiqc.log
│ │ │ └── multiqc_sources.txt
│ │ └── multiQC_report_BAMs_dedup.html
│ ├── flagstat
│ │ ├── sample_flagstat_filtered_bam_woMT_dedup.txt
│ │ └── sample_flagstat_woMT_dedup_shifted_sorted.txt
│ ├── fragmentSizeDistribution_plots
| | ├── ALL.samples_fragment_size_distribution.pdf
│ │ └── sample_fragment_size_distribution.pdf
│ ├── metrics
│ │ └── sample_metrics_woMT_dedup_bam.txt
│ └── unshifted_bams
│ ├── sample_mapQ20_sorted_woMT_dedup.bam
│ └── sample_mapQ20_sorted_woMT_dedup.bai
|
├── 04_Normalization
│ ├── HMCan_output ### --> only if HMCan correction is performed ###
│ └── normalized_bigWigs
│ └── sample_mapQ20_woMT_dedup_shifted_normalized_bs5.bw
│
├── 05_Peaks_MACS3 ### --> if HMCan correction is not performed ###
│ ├── sample_mapQ20_woMT_dedup_shifted_FDR0.01_peaks.narrowPeak
│ ├── sample_mapQ20_woMT_dedup_shifted_FDR0.01_peaks.xls
│ ├── sample_mapQ20_woMT_dedup_shifted_FDR0.01_summits.bed
│ └── log
│ └── sample_mapQ20_woMT_dedup_shifted_FDR0.01.log
|
├── 06_Overall_quality_and_info
| ├── Lorenz_curve_deeptools.plotFingreprint_allSamples.pdf
| ├── Counts
| │ ├── counts_summary.txt
| │ └── subread_featureCounts_output
| │ └── sample
| │ ├── sample.readCountInPeaks
| │ ├── sample.readCountInPeaks.log
| │ └── sample.readCountInPeaks.summary
| └── Sample_comparisons
| ├── multiBigWigSummary_matrix_allSamples.npz
| ├── PCA_on_BigWigs_wholeGenome.pdf
| ├── Peak_comparison
| │ ├── all_samples_peaks_concatenation_collapsed_sorted.bed
| │ ├── peaks_score_matrix_all_samples_MACS3.npz
| │ └── peaks_score_matrix_all_samples_table_MACS3.tsv
| | └── Heatmaps
| | ├── Heatmap_on_log1p.rawScores_for_MACS3.peaks_union_population.pdf
| │ └── Heatmap_on_zScores_for_MACS3.peaks_union_population.pdf
| └── Sample_correlation
| ├── Correlation_heatmap_on_BigWigs_wholeGenome_pearsonMethod.pdf
| ├── Correlation_heatmap_on_BigWigs_wholeGenome_spearmanMethod.pdf
| ├── Correlation_scatterplot_on_BigWigs_wholeGenome_pearsonMethod.pdf
| └── Correlation_scatterplot_on_BigWigs_wholeGenome_spearmanMethod.pdf
|
└── 07_Variant_calling
├── all_samples_peaks_concatenation_collapsed_sorted.bed
├── all.samples_dedup_gatk-indel_filtered.DP20.QUAL20
├── all.samples_dedup_gatk-snp_filtered.DP20.QUAL20
├── all.samples_INDEL_counts_plot.pdf
├── all.samples_SNP_counts_plot.pdf
└── sample
├── sample_dedup_gatk-indel_filtered.DP20.QUAL20.txt
├── sample_dedup_gatk-indel_filtered.DP20.QUAL20.vcf.gz
├── sample_dedup_gatk-indel_filtered.DP20.QUAL20.vcf.gz.tbi
├── sample_dedup_gatk-snp_filtered.DP20.QUAL20.txt
├── sample_dedup_gatk-snp_filtered.DP20.QUAL20.vcf.gz
├── sample_dedup_gatk-snp_filtered.DP20.QUAL20.vcf.gz.tbi
├── sample_dedup_gatk.vcf.gz
├── sample_dedup_gatk.vcf.gz.tbi
├── sample_mapQ20_sorted_woMT_dedup_bsqr.bai
├── sample_mapQ20_sorted_woMT_dedup_bsqr.bam
├── sample_mapQ20_sorted_woMT_dedup_bsqr.table
└── sample_plotCoverage.pdf
01_fastQC_raw
This folder contains a the fastq quality control (fastQC) reports for each fastq file and a summary report of multiQC.
02_BAM
When the reads are aligned onto the reference genome by bwa, the resulting SAM files are filtered for mapping quality (MAPQ) and the mithocondrial (suffix: woMT) reads are removed before sorting. Flagstat metrics is generated for each file and stored in the homonym folder.
03_BAM_dedup / 03_BAM_mdup
PICARD is used to remove (suffix: dedup) or mark (suffix: mdup) duplicates in the BAM files. The resulting BAMs are stored in the subfolder “unshifted_bams”, while the PICARD metrics is stored in the “metrics” folder. A fastq quality control (fastQC) and relative multiQC report is performed on the unshifted bams.
Then, the Tn5 nick reparation bias is corrected by shifting of the reads using deeptools alignmentSieve (suffix: shifted). Notice that, after shifting, the read sequence information is lost in the shifted BAMs.
Flagstat metrics is generated for each unshifted and shifted bam file and stored in the “falgstat” folder.
Furthermore, in the “fragmentSizeDistribution_plots” folder the distribution of the fragment sizes for each sample (shifted BAMs) and a file collecting all the plots in a single file. Here after an example of a good (left) and a bad (right) fragment size distribution.

An optimal fragment size distribution should be included within a range of 50-800bp, with a periodicity of ~150bp (corrsponding to mono-, di-, tri-, … nucleosomes) with a lower intensity for larger fragments.
04_Normalization
Shifted signal is normalized on sequencing depth library upon copy number variation correction by HMCan (if requested by the user). The bin size used is indicated in the resulting bigWig file name (suffix: bs#).
However, these bigWig files can be normalized more precisely normalized in the case that you dispone of a corresponding RNA-seq data set using CHIPIN (L. Polit et.al, BMC Bioinformatics, 2021). Examples of CHIPIN usage can be found at S. Gregoricchio et al., Nucleic Acids Research (2022).
05_Peaks_MACS3 (when HMCan correction is not performed)
Peaks and summits (if required by the user) are called by MACS3 on shifted BAMs. The FDR (False Discovery Ratio) threshold used is indicated in the file name (suffix: FDR#). When HMCan correction is active, the peaks are called by HMCan itself.
06_Overall_quality_and_info
This folder contains multiple quality controls, feature counts and sample correlation plots:
Lorenz_curve_deeptools.plotFingreprint_allSamples.pdfis a plot showing the enrichment of the signal allover the genome. Indeed, if a sample does not show any enrichment the reads will equally distributed over the genome resulting in a diagonal line in the plot (left panel). When instead the signal is specific for the feature sought (e.g., open chromatin) it will be enriched only at specific location and the curve will be closer to the bottom-right corner of the plot (right panel).

Counts: contains the results of featureCounts (from subread) with the counts of reads and other statistics on called peaks for each sample. It is availble also tab-separated file containing a summary of the main features counts for each sample:
Summary counts table description
| Column | Description |
|---|---|
| Sample | Sample name |
| Reads_R1 | Number of reads in read.1 fastq file. |
| Reads_R2 | Number of reads in read.2 fastq file. |
| Reads_total | Total number of reads (read.1 + read.2). |
| unfiltered_BAM | Total number of reads in the bam file after filtering by map quality (MAPQ). |
| Percentage_MT | Approximative percentage of reads represented by the mithocondrial DNA. Ideally lower than 10-20%. |
| dedup_BAM | Total number of reads left after BAM reads deduplication. |
| duplicated_reads | Number of duplicated reads. If the duplicates are not remove the value will be 0. |
| shifted_BAM | Number of reads in the shifted BAMs. |
| loss_post_shifting | Number of reads lost upon BAM shifting. Consider that reads falling in blacklisted regions are removed. |
| n.peaks | Total number of peaks called. |
| FRiP.perc | Frequency Reads in Peaks percentage, corresponds to the number of reads falling in peak regions divide by the total number of reads and multiplied by 100. |
| FRiP.quality | A label (“good” or “bad”) to indicate whether the FRiP score is good or not for a given sample. The threshold can be changed in the config file by the user, by the default 20 (as suggested by the ENCODE guidelines). |
Sample_comparisons: the plots in this folder help the user to understand the variability of the samples.multiBigWigSummary_matrix_allSamples.npz: result of deeptools multiBigWigSummary used to plot the PCA and correlation plots;PCA_on_BigWigs_wholeGenome.pdf: Principal Component Analyses results of the signal allover the genome;Peak_comparison:all_samples_peaks_concatenation_collapsed_sorted.bed: the peaks called in all samples are merged and collapsed in this bed file;peaks_score_matrix_all_samples_MACS3.npz: a matrix containing the average score at each peak (previous bed file) for each samples is generated;peaks_score_matrix_all_samples_table_MACS3.tsv: same matrix as before, but in tab-separated format.Heatmaps: the matrix generated on all peaks is used to cluster the samples and two heatmaps are plotted: one on the log1p of the raw scores, and one on the z-score (on rows)
Sample_correlation: scatter and heatmap correlation plots are generated based on the signal over the whole genome. Both Pearson and Spearman methods are used.
07_Variant_calling
If required by the user, the pipeline can call altered single-nucleotide polymorphism (SNP) and insertion/deletions (InDel). PICARD CreateSequenceDictionary is used to create a genome dictionary in order to perform a Base Quality Score Recalibration (BQSR) of unshifted BAM files (after filling of the ReadGroups by PICARD AddOrReplaceReadGroups) by GATK BaseRecalibrator and GATK ApplyBQSR.
The recalibrated BAMs are used by GATK HaplotypeCaller to generate a GVCF (Genomic Variant Call Format) file containing the genomic variants individuated at the regions included in the file resulting by the merge of all the called peaks. This GVCF file is then recalibrated by GATK GenotypeGVCFs resulting in a VCF file.
The VCF recalibrated file is selected by GATK SelectVariants to obtain two separate VCF files corresponding to SNPs and InDels.
Ultimately, these VCF are hard-filtered for sequencing depth (DP) and quality (QUAL) by SnpSift Filter and then exported in a txt table by SnpSift Extract Fields. Notably, the genotype ‘0|0’ (both alleles not mutated) is filtered out from the .txt table.
The SNP, or InDel, .txt tables from all samples are merged in a unique one with the addition of a column corresponding to the sample name. These tables are used to generate a plot of the counts of variants found in each sample.
Further, a coverage plot at the merged peaks is generated by deeptools plotCoverage.
Package history and releases
A list of all releases and respective description of changes applied could be found here.
Contact
For any suggestion, bug fixing, commentary please report it in the issues/request tab of this repository.
License
This repository is under a GNU General Public License (version 3).